Clustering Run Example
Clustering Run Example
In the example below, I conduct the run in the examples/clustered directory but there isn’t really anything there. It is just a space for conducting the analyses. With this example, we will conduct a clustering run for PyPHLAWD on the Adoxaceae plant clade.
Setting things up
We are going to assume that you have already installed all the dependencies (if not, head over to the installation instructions). This animation below starts from cloning the repo (again, assuming that all the dependencies are installed). This includes changing a couple parameters in the conf.py and compiling the cython file.

Commands from gif
git clone https://github.com/FePhyFoFum/PyPHLAWD.gitcd PyPHLAWD/srcbash compile_cython.sh- edit
conf.pychanging theDIto the directory where PyPHLAWDsrcis, changing thesmallest_size=500, and changinglength_limit=0.5 cd ../examples/clusteredpython ../../src/setup_clade.py Adoxaceae ~/Desktop/pln.041118.db . log.md.gz
Starting a run
We have the run going now (and the gif is a little sped up). But all of the commands can be found in the log.md.gz that is created. Check out the baited example here if you want to see what that file looks like.
Commands from gif
python ../../src/setup_clade.py Adoxaceae ~/Desktop/pln.041118.db . log.md.gz
Looking at the results
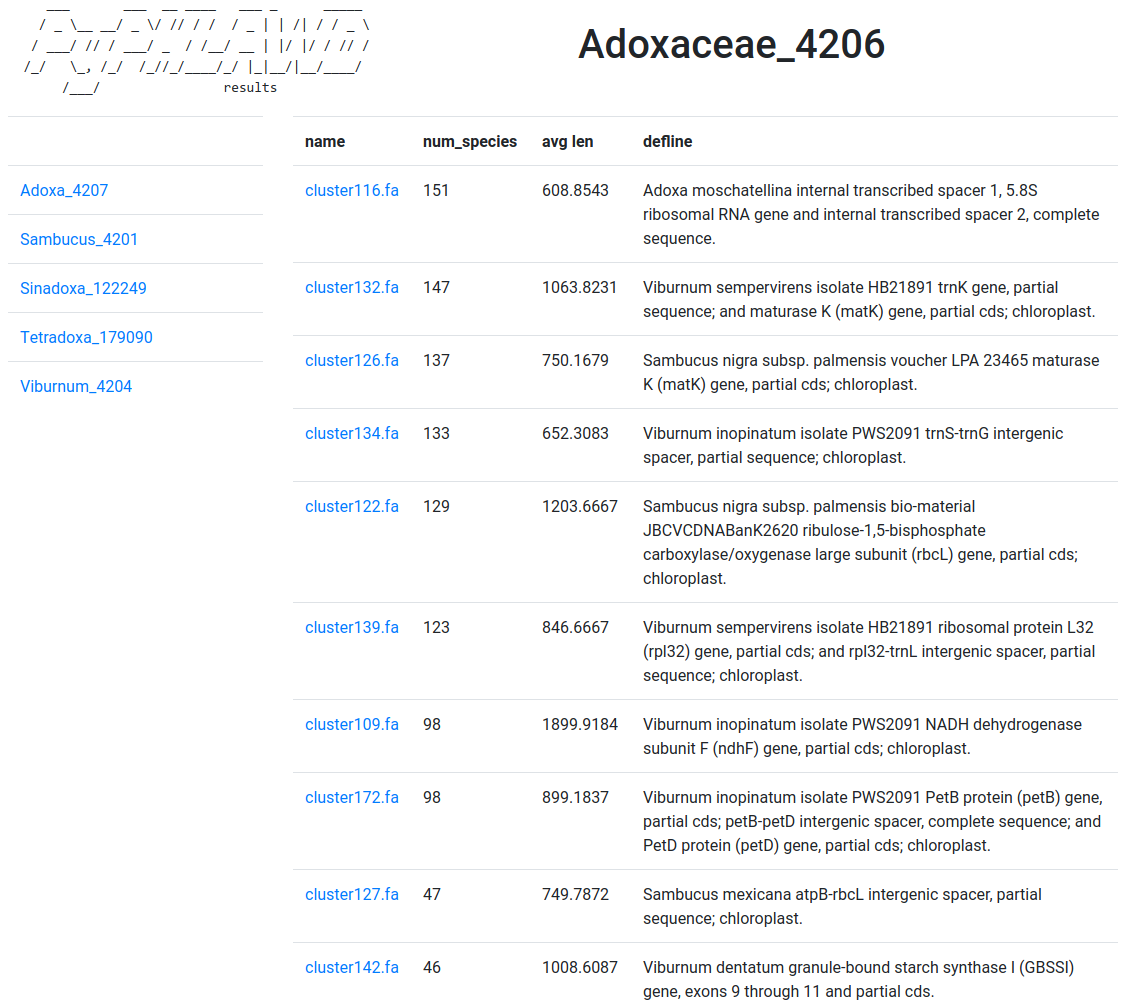
Inside the Adoxaceae_4206, there is a info.html file that you can see (part of) here. The clusters themselves can be found in Adoxaceae_4206/clusters.
Finding good clusters
You can manually pick what clusters you like or you can use a script called find_good_clusters_for_concat.py as demonstrated here. This script will rename the clusters from GenBank ids to NCBI taxon ids. If you want to manually do this, the script is called change_id_to_ncbi_fasta_mult.py and is demonstrated in the baited example here.
Commands from gif
python ../../src/find_good_clusters_for_concat.py Adoxaceae_4206/Do you want to rename these clusters? y/n/#yDo you want to make trees and trim tips for these gene regions y/nnDo you want to concat? y/nyDo you want to make a constraint? y/nn
Changing the names
I went ahead and conducted a phylogenetic analysis on the concatenated dataset from the above procedure. The resulting tree has NCBI taxon ids as the tip labels. I haven’t memorized these so I need taxon labels. To change that, you can use the script change_ncbi_to_name_tre.py as demonstrated below.
Commands from gif
python ../../src/change_ncbi_to_name_tre.py Adoxaceae_4206/Adoxaceae_4206.table Adoxaceae_4206/RAxML_bestTree.ADOX Adoxaceae_4206/RAxML_bestTree.ADOX.cn
What is in the directory
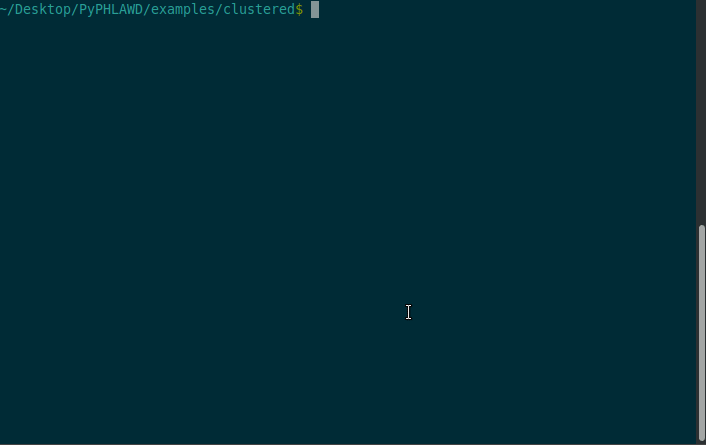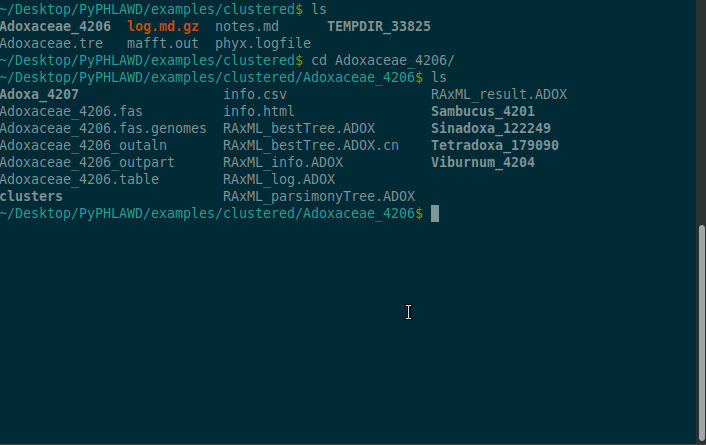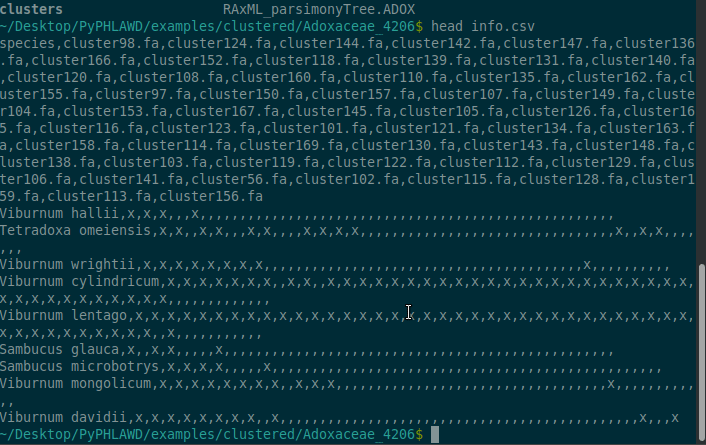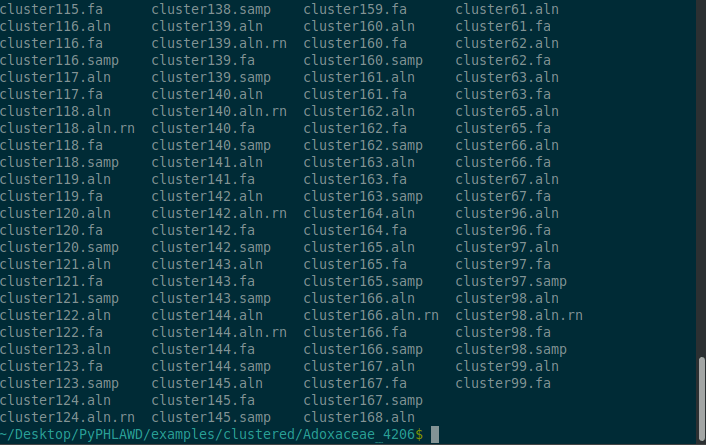
Here I am just showing some of the things in the directory. For example, the info.csv file has the occupancy matrix for the clusters.