
Bait Run Example
Bait Run Example
The files necessary to run this example can be found in the examples/baited directory. There is a baits directory with a few sequences (not comprehensive) for ITS, matK, rbcL, and trnLF. These files in the bait directory need to have .fa at the end of the filename. Other files will be skipped. With this example, we will conduct a baited run for PyPHLAWD on the Adoxaceae plant clade.
Setting things up
We are going to assume that you have already installed all the dependencies (if not, head over to the installation instructions). This animation below starts from cloning the repo (again, assuming that all the dependencies are installed).

Commands from gif
git clone https://github.com/FePhyFoFum/PyPHLAWD.gitcd PyPHLAWD/srcbash compile_cython.sh- edit
conf.pychanging theDIto the directory cd ../examples/baitedpython ../../src/setup_clade_bait.py Adoxaceae baits/ ~/Desktop/pln.041118.db . log.md.gz
Starting a run
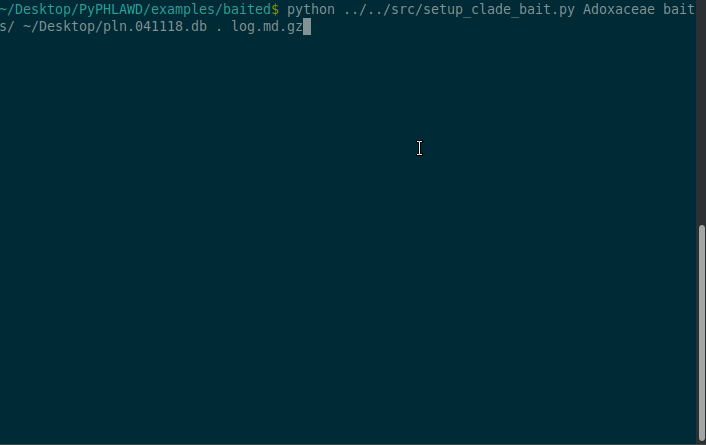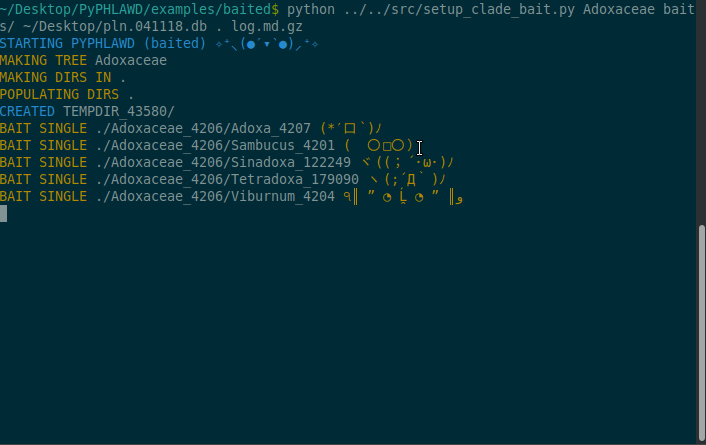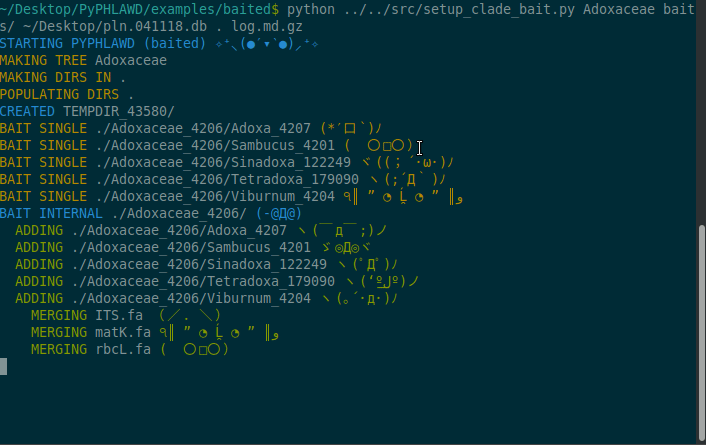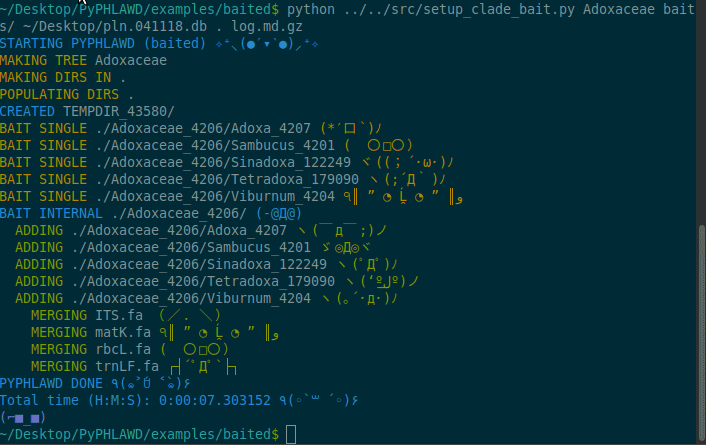
Now we are going to start a baited run using the bait in the baits directory in the examples/baited directory. As stated above, these files in the baits directory need to have .fa at the end of the filename. For this example, we are going to conduct the analysis on Adoxaceae. We need to have a database of the plant sequences from GenBank (constructed using phlawd_db_maker or downloaded from another source). We also provide a directory to put the results (the . refers to the current directory). Finally, we give an output filename (here, log.md.gz). The log file will be gzipped so that it isn’t too big and in a markdown format with each command that is run recorded.
Commands from gif
python ../../src/setup_clade_bait.py Adoxaceae baits/ ~/Desktop/pln.041118.db . log.md.gz
This is what the log file looks like in vim, which automatically ungzips the file for reading. If you want to look in another text editor, you can run gunzip log.md.gz that will make a file called log.md and then you can look at that file in whatever editor you like.
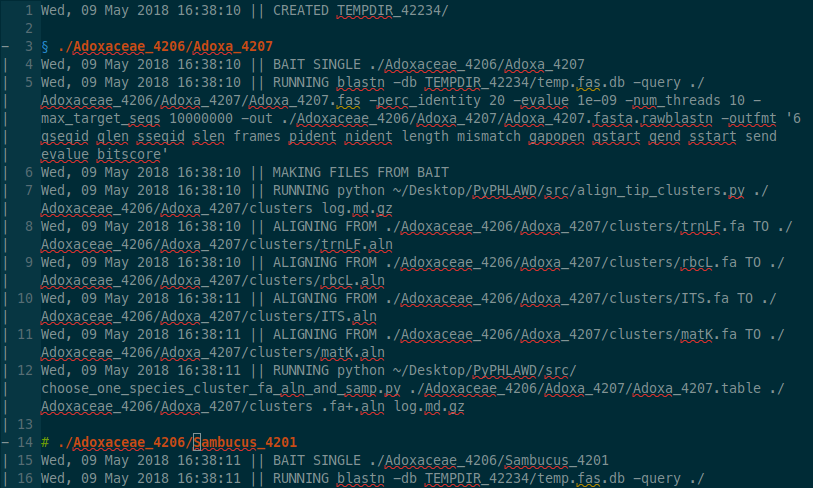
Changing the params
There are several parameters that you can change that might improve your results. Here, we are changing the length limit and the overlap parameters in the conf.py file. Remember that if you git pull PyPHLAWD, it will overwrite those changes, or you will need to stash them.
Commands from gif
- edit
../../src/conf.pyin a plain text editor (BBEdit, geany, vim, etc.) - change
smallest_sizeto450 - change
length_limitto0.5 python ../../src/setup_clade_bait.py Adoxaceae baits/ ~/Desktop/pln.041118.db . log.md.gz
Looking at the results
Inside the Adoxaceae_4206 that is created, there will be an info.html. Below are the results of that. If you would like to use the resulting clusters, they can be found in the Adoxaceae_4206/clusters directory.
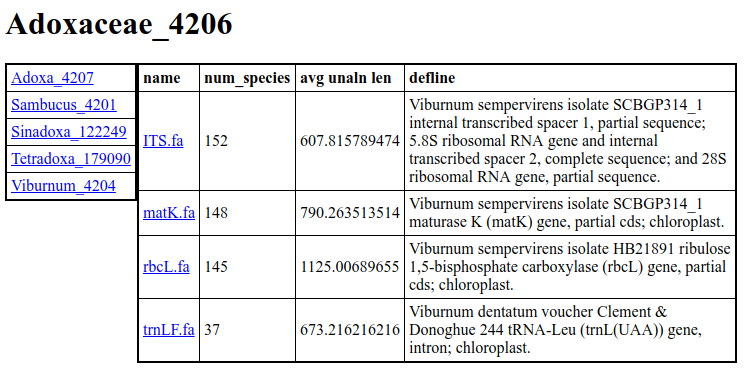
Preparing the files for analyses
The files that are found in Adoxaceae_4206/clusters directory include unaligned and aligned fasta files with ids corresponding to GenBank IDs. Below we should how you can change those to NCBI taxon ids (e.g., so they can be concatenated).
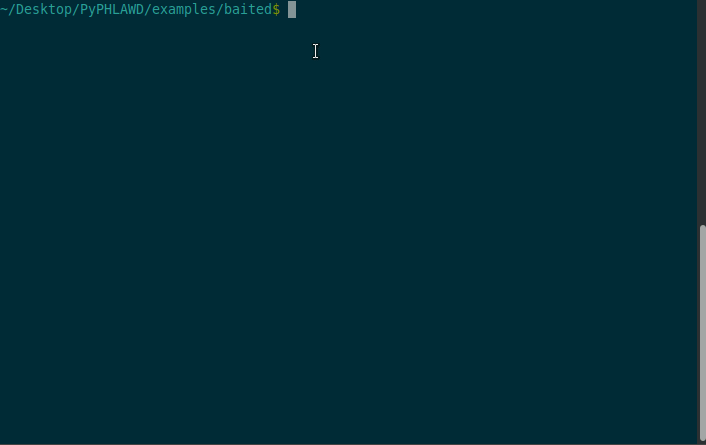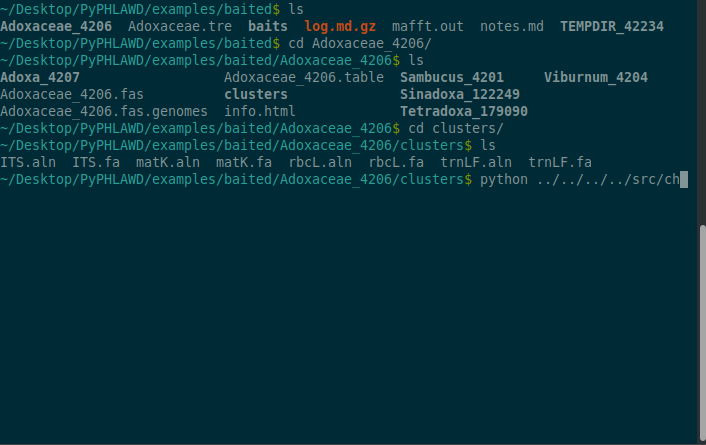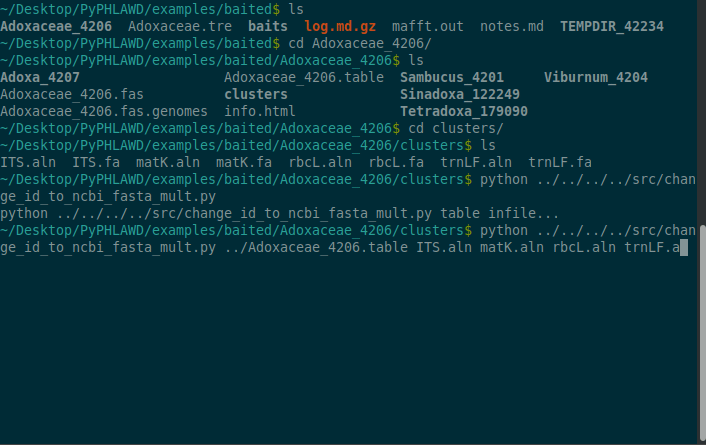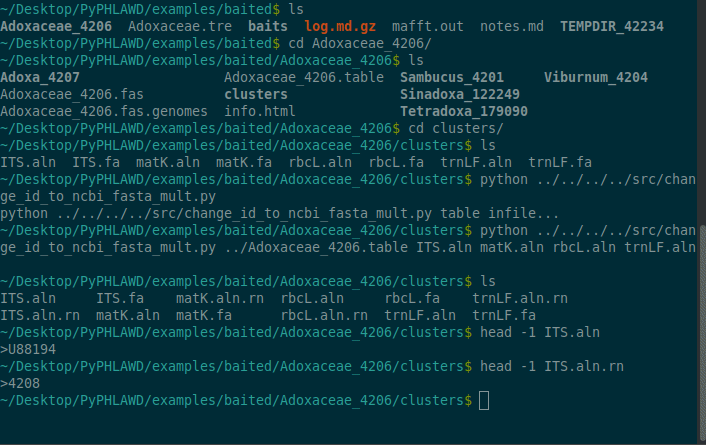
Commands from gif
cd Adoxaceae_4206python ../../../../src/change_id_to_ncbi_fasta_mult.py ../Adoxaceae_4206.table ITS.aln matK.aln rbcL.aln trnLF.aln
You can see the clustering analyses here.